First-principles study of order-disorder transitions in pseudo-binary clathrates
J. Brorsson,
A. E. C. Palmqvist,
and
P. Erhart
The Journal of Physical Chemistry C 125, 22817
(2021)
doi: 10.1021/acs.jpcc.1c06638
Download PDF
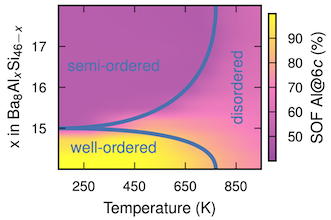
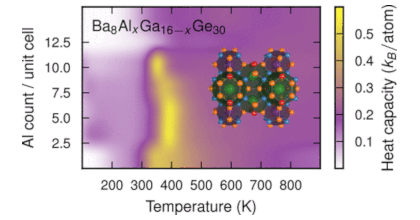
It has been recently demonstrated that the pseudo-ternary Ba8AlxGayGe46-x-y clathrate undergoes an order-disorder transition with increasing temperature that can be observed via the site occupation factors (SOFs) and manifests itself, e.g., in the electrical transport properties. Here, we generalise this result and analyse the characteristics of this order-disorder transition in the pseudo-binary clathrates Ba8GaxGe46-x, Ba8GaxSi46-x, Ba8AlxGe46-x, and Ba8AlxSi46-x. To this end, we employ atomistic simulations that combine alloy cluster expansions trained against density functional theory calculations with Wang-Landau and ensemble Monte Carlo simulations. The simulations show that all four systems studied here display order-disorder transitions for at least some composition range. Based on an extensive literature survey, we also provide evidence for signatures of the transition in earlier experimental studies that to the best of our knowledge have hitherto not been related to such transitions. The predicted transition temperatures are lower for Ba8GaxGe46-x and Ba8GaxSi46-x, than for Ba8AlxGe46-x and Ba8AlxSi46-x, although it appears that the simulations underestimate the transition temperatures for Ga-containing systems compared to experiment. This nonetheless provides a sensible explanation for why the experimentally determined Al SOFs agree better with the simulated high-temperature disordered configurations while the Ga SOFs more closely agree with the simulated ground state configurations. As a result of stronger interactions, the SOFs vary substantially especially near the stoichiometric 16:30 composition, providing an indication why it has proven difficult to synthesise Ba8AlxGe46-x and Ba8AlxSi46-x samples at this ratio. The present study thereby yields detailed atomic scale insight into the ordering in inorganic clathrates that, given the connection to transport properties established earlier, is not only useful from a fundamental perspective but also relevant for applications.