Understanding chemical ordering in intermetallic clathrates from atomic scale simulations
M. Ångqvist
and
P. Erhart
Chemistry of Materials 29, 7554
(2017)
doi: 10.1021/acs.chemmater.7b02686
Download PDF
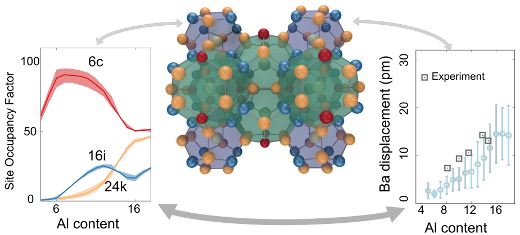
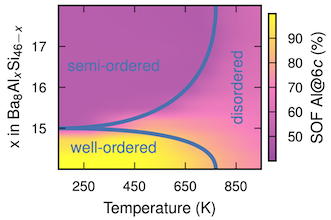
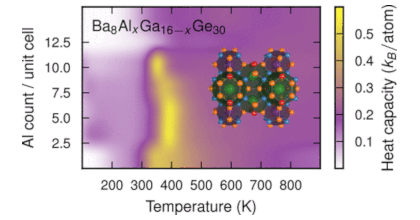
Intermetallic clathrates exhibit great variability with respect to elemental composition and distribution. While this provides a lot of flexibility for tuning properties it also poses a challenge with regard to developing a comprehensive understanding of these systems. Here, we employ a combination of alloy cluster expansions and density functional theory calculations to exhaustively sample the compositional space with ab-initio accuracy. We apply this methodology to study chemical ordering and related properties in the clathrate systems Ba8AlxSi46−x, Ba8AlxGe46−x, Ba8GaxGe46−x, and Ba8GaxSi46−x as a function of composition and temperature. We achieve very good agreement with the available experimental data for the site occupancy factors (SOFs) even for stoichiometries outside the composition range considered during construction of the cluster expansions. This validation enables us to reconcile the variations in the experimental data and explain non-monotonic variations of the SOFs. In particular, we provide a rationale for the extreme SOF behavior with varying composition observed in Al based clathrates. Furthermore, we quantify the effect of chemical ordering on both heat capacity and lattice expansion. Finally, we determine the effect of chemical disorder on the displacements of the guest species (Ba), which enables us to at least partially explain experimental observations of the nuclear density of Ba in different clathrates.
Associated data
Structures obtained by simulated annealing Permalink (ZIP archive containing xyz-files)