Optimization of the Thermoelectric Power Factor: Coupling between Chemical Order and Transport Properties
M. Ångqvist,
D. O. Lindroth,
and
P. Erhart
Chemistry of Materials 28, 6877
(2016)
doi: 10.1021/acs.chemmater.6b02117
Download PDF
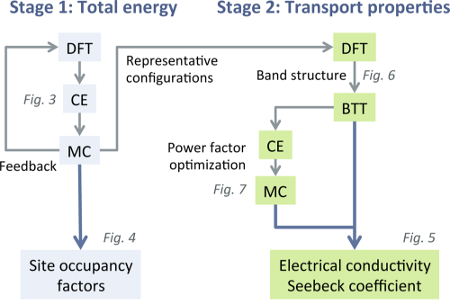
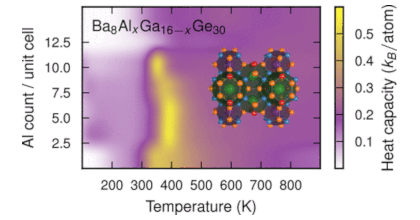
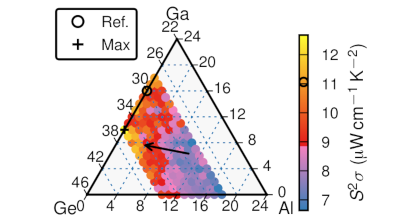
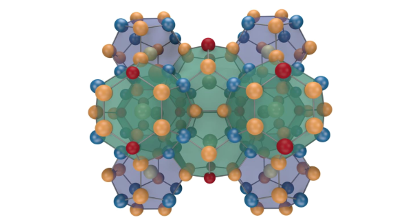
Many thermoelectric materials are multicomponent systems that exhibit chemical ordering, which can affect both thermodynamic and transport properties. Here, we address the coupling between order and thermoelectric performance in the case of a prototypical inorganic clathrate (Ba8Ga16Ge30) using a combination of density functional and Boltzmann transport theory as well as alloy cluster expansions and Monte Carlo simulations. The calculations describe the experimentally observed site occupancy factors and reproduce experimental data for the transport coefficients. By inverting the cluster expansion, we demonstrate that the power factor can be increased by more than 60% for certain chemical ordering patterns that involve reducing the number of the trivalent species on the 6c Wyckoff site. This enhancement is traced to specific features of the electronic band structure. The approach taken in the present work can be readily adapted to other materials and enables a very general form of band structure engineering. In this fashion, it can guide the computational design of compounds with optimal transport properties.
Associated data
Ground state configuration Permalink (CIF file) Power-factor-optimized structure Permalink (CIF file)