Hydrogen-driven Surface Segregation in Pd-alloys from Atomic Scale Simulations
P. Ekborg-Tanner
and
P. Erhart
The Journal of Physical Chemistry C 125, 17248
(2021)
doi: 10.1021/acs.jpcc.1c00575
zenodo: 5076169
(associated data)
Download PDF
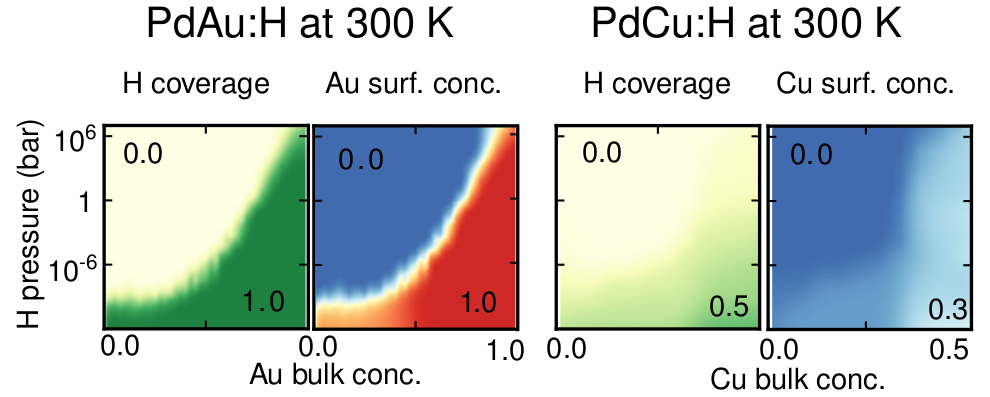
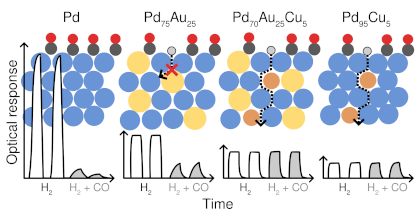
The safe widespread application of hydrogen-based fuels requires sensors that are long-term stable, inexpensive, hydrogen specific and have a short response time. In this regard, optical sensing based on nanostructured Pd-alloys has shown great potential but challenges remain, including understanding and controlling the surface composition under operation conditions. While the latter is crucial for long-term functionality and stability, it is experimentally very challenging to obtain accurate atomic-scale information. Here, we therefore scrutinize the behavior of two particularly relevant surface alloys, 111 AuPd and 111 CuPd, in H environments ranging from vacuum to fully covered surfaces. To this end, we employ a combination of alloy cluster expansions trained using density functional theory calculations, Monte Carlo simulations and thermodynamic analysis to obtain the H coverage as well as the layer-by-layer composition of the near-surface region as a function of H partial pressure, temperature and bulk composition. To overcome the symmetry reduction implicit to surface systems, we exploit local symmetries, which enables us to achieve accurate and reliable models at a small computational cost. In the case of AuPd, Au segregates to the surface in vacuum while Pd segregates to the surface at 100% H coverage, and the transition between these regimes occurs over a narrow H pressure interval. In the case of CuPd, on the other hand, the H coverage varies much more gradually with H pressure and is coupled to a non-monotonic variation of the Cu concentration in the topmost surface layer. While there is a pronounced Cu depletion both at 0 and 100% H coverage, Cu enrichment is observed at 50% coverage at Cu bulk concentrations up to at least 10%, providing a non-trivial explanation for an apparent discrepancy between experiment and calculations that was observed previously. At the same time, layer 2 continuously shifts from Cu enrichment to Cu depletion with increasing H coverage. The results demonstrate the rich behavior that can result from the coupling of metal-metal and metal-hydrogen interactions at surfaces, even in apparently simple but concentrated systems. Moreover, they underline the advantages of simulations that account for temperature and pressure effects as well as models that can accurately capture the interactions over a wide composition range.