General-purpose machine-learned potential for 16 elemental metals and their alloys
K. Song,
R. Zhao,
J. Liu,
Y. Wang,
E. Lindgren,
Y. Wang,
S. Chen,
K. Xu,
T. Liang,
P. Ying,
N. Xu,
Z. Zhao,
J. Shi,
J. Wang,
S. Lyu,
Z. Zeng,
S. Liang,
H. Dong,
L. Sun,
Y. Chen,
Z. Zhang,
W. Guo,
P. Qian,
J. Sun,
P. Erhart,
T. Ala-Nissila,
Y. Su,
and
Z. Fan
arXiv:2311.04732
doi: 10.48550/arXiv.2311.04732
zenodo: 10081677
(associated data)
Download PDF
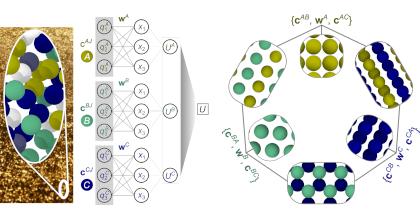
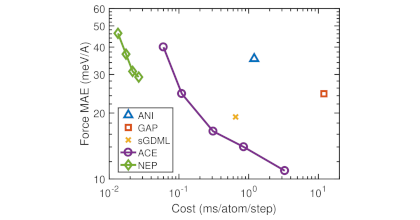
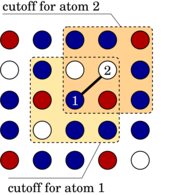
Machine-learned potentials (MLPs) trained against quantum-mechanical reference data have demonstrated remarkable accuracy, surpassing empirical potentials. However, the absence of readily available general-purpose MLPs encompassing a broad spectrum of elements and their alloys hampers the applications of MLPs in materials science. In this study, we present a feasible approach for constructing a unified general-purpose MLP for numerous elements and showcase its capability by developing a model (UNEP-v1) for 16 elemental metals (Ag, Al, Au, Cr, Cu, Mg, Mo, Ni, Pb, Pd, Pt, Ta, Ti, V, W, Zr) and their diverse alloys. To achieve a complete representation of the chemical space, we demonstrate that employing 16 one-component and 120 two-component systems suffices, thereby avoiding the enumeration of all 65 535 possible combinations for training data generation. Furthermore, we illustrate that systems with more components can be adequately represented as interpolation points in the descriptor space. Our unified MLP exhibits superior performance across various physical properties as compared to the embedded-atom method potential, while maintaining computational efficiency. It achieves a remarkable computational speed of 1.5× 108</sup atom· step/second in molecular dynamics simulations using eight 80-gigabyte A100 graphics cards, enabling simulations up to 100 million atoms. We demonstrate the generality and high efficiency of the MLP in studying plasticity and primary radiation damage in the MoTaVW refractory high-entropy alloys, showcasing its potential in unraveling complex materials behavior. This work represents a significant leap towards the construction of a unified general-purpose MLP encompassing the periodic table, with profound implications for materials research and computational science.