Composition-dependent interatomic potentials: A systematic approach to modelling multicomponent alloys
B. Sadigh,
P. Erhart,
A. Stukowski,
and
A. Caro
Philosophical Magazine 89, 3371
(2009)
doi: 10.1080/14786430903292373
Download PDF
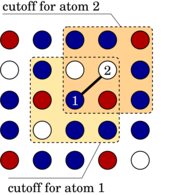
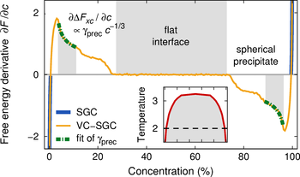
We propose a simple scheme to construct composition-dependent interatomic potentials for multicomponent systems that, when superposed onto the potentials for the pure elements, can reproduce not only the heat of mixing of the solid solution in the entire concentration range but also the energetics of a wider range of configurations including intermetallic phases. We show that an expansion in cluster interactions provides a way to systematically increase the accuracy of the model, and that it is straightforward to generalise this procedure to multicomponent systems. Concentration-dependent interatomic potentials can be built upon almost any type of potential for the pure elements including embedded atom method (EAM), modified EAM, bond-order, and Stillinger-Weber type potentials. In general, composition-dependent N-body terms in the total energy lead to explicit (N+1)-body forces, which potentially render them computationally expensive. We present an algorithm that overcomes this problem and that can speed up the calculation of the forces for composition-dependent pair potentials in such a way as to make them computationally comparable in efficiency and scaling behaviour to standard EAM potentials. We also discuss the implementation in Monte Carlo simulations. Finally, we exemplarily review the composition-dependent EAM model for the Fe-Cr system.