Efficient implementation of the concentration-dependent embedded atom method for molecular dynamics and Monte-Carlo simulations
A. Stukowski,
B. Sadigh,
P. Erhart,
and
A. Caro
Modelling and Simulation in Materials Science and Engineering 17, 31237
(2009)
doi: 10.1088/0965-0393/17/7/075005
Download PDF
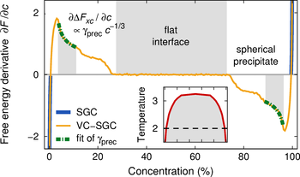
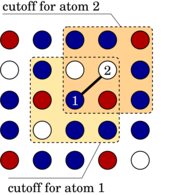
The concentration-dependent embedded atom method (CD-EAM) is a powerful model for atomistic simulation of concentrated alloys with arbitrarily complex mixing enthalpy curves. In this paper, we show that in spite of explicit three-body forces, this model can be implemented quite simply with a computational efficiency comparable to the standard EAM for molecular-dynamics (MD) simulations. Ready-to-use subroutines for the parallel MD code LAMMPS can be provided by the authors upon request. We further propose an improved version of this potential that allows for very efficient calculations of single-particle displacement/transmutation energies, while retaining the complexity implicit in the three-body interactions. This enables large-scale Monte-Carlo simulations of alloys with the interatomic interactions described by the CD-EAM model.