The hiphive package for the extraction of high-order force constants by machine learning
F. Eriksson,
E. Fransson,
and
P. Erhart
Advanced Theory and Simulations 2, 1800184
(2019)
arXiv:1811.09267
doi: 10.1002/adts.201800184
zenodo: 7938851
(associated data)
Download PDF
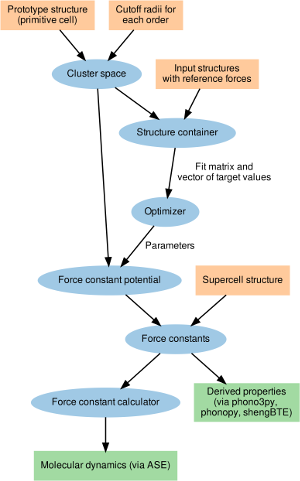
The efficient extraction of force constants (FCs) is crucial for the analysis of many thermodynamic materials properties. Approaches based on the systematic enumeration of finite differences scale poorly with system size and can rarely extend beyond third order when input data is obtained from first-principles calculations. Methods based on parameter fitting in the spirit of interatomic potentials, on the other hand, can extract FC parameters from semi-random configurations of high information density and advanced regularized regression methods can recover physical solutions from a limited amount of data. Here, we present the hiphive Python package, that enables the construction of force constant models up to arbitrary order. hiphive exploits crystal symmetries to reduce the number of free parameters and then employs advanced machine learning algorithms to extract the force constants. Depending on the problem at hand both over and underdetermined systems are handled efficiently. The FCs can be subsequently analyzed directly and or be used to carry out e.g., molecular dynamics simulations. The utility of this approach is demonstrated via several examples including ideal and defective monolayers of MoS2 as well as bulk nickel.