Efficient calculation of the lattice thermal conductivity by atomistic simulations with ab-initio accuracy
J. Brorsson,
A. Hashemi,
Z. Fan,
E. Fransson,
F. Eriksson,
T. Ala-Nissila,
A. V. Krasheninnikov,
H. Komsa,
and
P. Erhart
Advanced Theory and Simulations 4, 2100217
(2021)
doi: 10.1002/adts.202100217
zenodo: 5034182
(associated data)
Download PDF
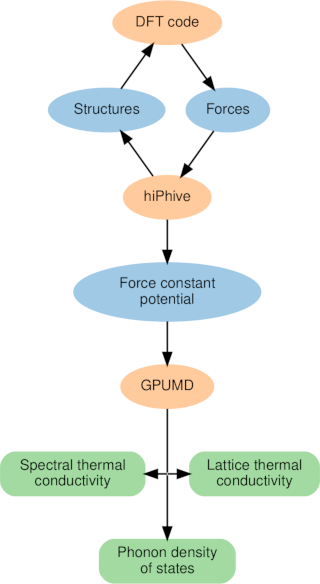
High-order force constant expansions can provide accurate representations of the potential energy surface relevant to vibrational motion. They can be efficiently parametrized using quantum mechanical calculations and subsequently sampled at a fraction of the cost of the underlying reference calculations. Here, we combine force constant expansions via the hiphive package with GPU-accelerated molecular dynamics simulations via the gpumd package to obtain an accurate, transferable and efficient approach for sampling the dynamical properties of materials. We demonstrate the performance of this methodology by applying it both to materials with very low thermal conductivity (Ba8Ga16Ge30, SnSe) and a material with a relatively high lattice thermal conductivity (monolayer-MoS2). These cases cover both situations with weak (monolayer-MoS2, SnSe) and strong (Ba8Ga16Ge30) phonon renormalization. The simulations also enable us to access complementary information such as the spectral thermal conductivity, which allows us to discriminate the contribution by different phonon modes while accounting for scattering to all orders. The software packages described here are made available to the scientific community as free and open-source software in order to encourage the more widespread use of these techniques as well as their evolution through continuous and collaborative development.

