Modeling the electrical conductivity in BaTiO3 on the basis of first-principles calculations
P. Erhart
and
K. Albe
Journal of Applied Physics 104, 18637
(2008)
doi: 10.1063/1.2956327
Download PDF
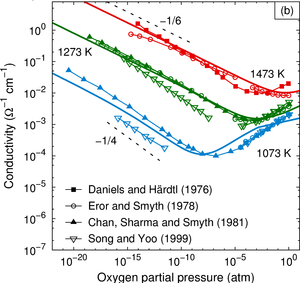
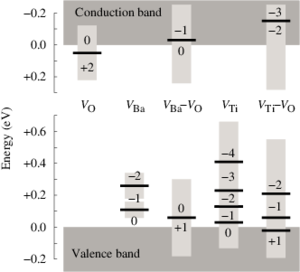
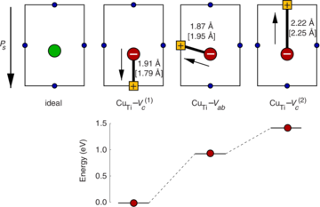
The dependence of the electrical conductivity on the oxygen partial pressure is calculated for the prototypical perovskite BaTiO3 based on data obtained from first-principles calculations within density functional theory. The equilibrium point defect concentrations are obtained via a self-consistent determination of the electron chemical potential. This allows one to derive charge carrier concentrations for a given temperature and chemical environment and eventually the electrical conductivity. The calculations are in excellent agreement with experimental data if an accidental acceptor dopant level of 1017 cm3 is assumed. It is shown that doubly charged oxygen vacancies are accountable for the high-temperature n-type conduction under oxygen-poor conditions. The high-temperature p-type conduction observed at large oxygen pressures is due to barium vacancies and titanium-oxygen divacancies under Ti- and Ba-rich conditions, respectively. Finally, the connection between the present approach and the mass-action law approach to point defect thermodynamics is discussed.