Intrinsic point defects in aluminum antimonide
D. Åberg,
P. Erhart,
A. J. Williamson,
and
V. Lordi
Physical Review B 77, 165206
(2008)
doi: 10.1103/PhysRevB.77.165206
Download PDF
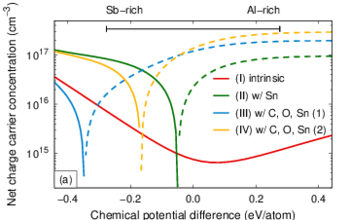
Calculations within density functional theory on the basis of the local density approximation are carried out to study the properties of intrinsic point defects in aluminum antimonide. Special care is taken to address finite-size effects, band gap error, and symmetry reduction in the defect structures. The correction of the band gap is based on a set of GW calculations. The most important defects are identified to be the aluminum interstitial Ali,Al(1+), the antimony antisites SbAl(0) and SbAl(1+), and the aluminum vacancy VAl(3-). The intrinsic defect and charge carrier concentrations in the impurity-free material are calculated by self-consistently solving the charge neutrality equation. The impurity-free material is found to be n-type conducting at finite temperatures.