Thermodynamics of mono- and di-vacancies in barium titanate
P. Erhart
and
K. Albe
Journal of Applied Physics 102, 084111
(2007)
doi: 10.1063/1.2801011
Download PDF
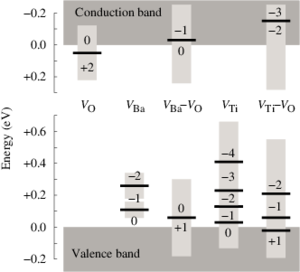
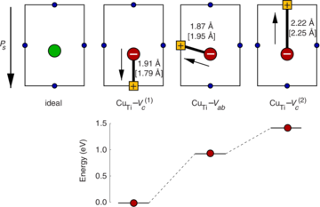
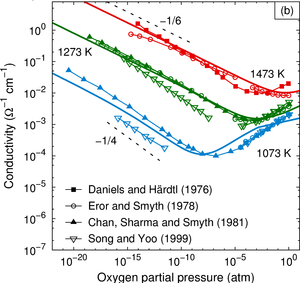
The thermodynamic and kinetic properties of mono- and di-vacancy defects in cubic (para-electric) barium titanate BaTiO3 are studied by means of density-functional theory calculations. It is determined which vacancy types prevail for given thermodynamic boundary conditions. The calculations confirm the established picture that vacancies occur in their nominal charge states almost over the entire band gap. For the dominating range of the band gap the di-vacancy binding energies are constant and negative. The system, therefore, strives to achieve a state in which, under metal-rich (oxygen-rich) conditions, all metal (oxygen) vacancies are bound in di-vacancy clusters. The migration barriers are calculated for mono-vacancies in different charge states. As oxygen vacancies are found to readily migrate at typical growth temperatures, di-vacancies can be formed at ease. The key results of the present study with respect to the thermodynamic behavior of mono- and di-vacancies influence the initial defect distribution in the ferroelectric phases and therefore the conditions for aging.