Machine learning models for molecular potential energy surfaces
V. Martvall
Master′s Thesis
(2022)
doi: 20.500.12380/304815
Download PDF

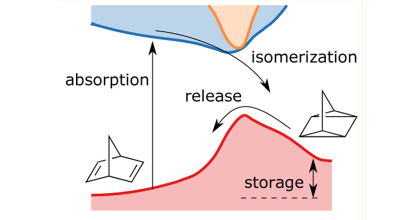
Molecular solar thermal storage (MOST) systems is a type of energy storage system which consist of photo switchable molecules that can convert solar energy into chemical energy which can be released on demand. One important property of a MOST system is that it the energy storage time should be long which means that the so-called back-conversion barrier should be large. Recently an experimental study showed that a substitution in the ortho position of the molecular photo switch norbornadiene leads to a significant increase of the back-conversation barrier. However in the same study the back-conversion barrier was also investigated with electronic structure calculations and this behavior was not seen. This thesis aimed to understand this discrepancy by investigating the temperature dependence of the back-conversion for two derivatives of norbornadiene by combining a machine learning (ML) method called symmetric gradient domain machine learning (sGDML) with electronic structure calculations to obtain computationally efficient models describing the dynamical landscape of the molecules studied. It was seen that sGDML models could describe the dynamical landscape of such molecules to a good approximation, however more work is required to obtain models which are accurate enough to to study the temperature dependence of the back-conversion.