A Comparative Ab-Initio Study of Substituted Norbornadiene-Quadricyclane Compounds for Solar Thermal Storage
M. J. Kuisma,
A. M. Lundin,
K. Moth-Poulsen,
P. Hyldgaard,
and
P. Erhart
The Journal of Physical Chemistry C 120, 3635
(2016)
doi: 10.1021/acs.jpcc.5b11489
Download PDF
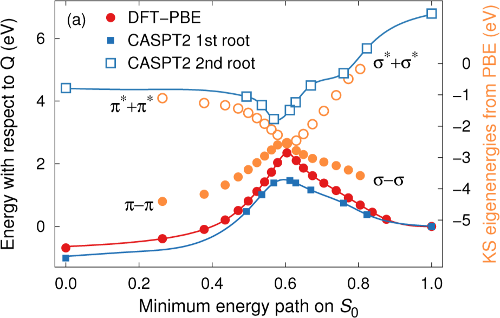
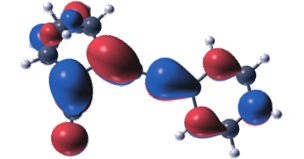
Molecular photoswitches that are capable of storing solar energy, so-called molecular solar thermal storage systems, are interesting candidates for future renewable energy applications. In this context, substituted norbornadiene-quadricyclane systems have received renewed interest due to recent advances in their synthesis. The optical, thermodynamic, and kinetic properties of these systems can vary dramatically depending on the chosen substituents. The molecular design of optimal compounds therefore requires a detailed understanding of the effect of individual substituents as well as their interplay. Here, we model absorption spectra, potential energy storage, and thermal barriers for back-conversion of several substituted systems using both single-reference (density functional theory using PBE, B3LYP, CAM-B3LYP, M06, M06-2x, and M06-L functionals as well as MP2 calculations) and multi-reference methods (complete active space techniques). Already the diaryl substituted compound displays a strong red-shift compared to the unsubstituted system, which is shown to result from the extension of the conjugated ?-system upon substitution. Using specific donor/acceptor groups gives rise to a further albeit relatively smaller red-shift. The calculated storage energy is found to be rather insensitive to the specific substituents, although solvent effects are likely to be important and require further study. The barrier for thermal back-conversion exhibits strong multi-reference character and as a result is noticeably correlated with the red-shift. Two possible reaction paths for the thermal back-conversion of diaryl substituted quadricyclane are identified and it is shown that among the compounds considered the path via the acceptor side is systematically favored. Finally, the present study establishes the basis for high throughput screening of norbornadiene-quadricyclane compounds as it provides guidelines for the level of accuracy that can be expected for key properties from several different techniques.