Atomic scale modeling of ordering phenomena
M. Ångqvist
Doctoral Thesis
(2020)
url: https://research.chalmers.se/publication/515685
Download PDF

Ordering phenomena in materials often have a crucial impact on materials properties. They are governed by the competition between entropy and energy. Accordingly simulating these aspects requires the construction of models that enable a computationally efficient exploration of the relevant configuration space. The alloy cluster expansion technique is particular well suited for this task as they can be trained to reach high accuracy while being computationally suitable for rapid sampling via Monte Carlo simulations.
In paper I we present the icet software for the construction and sampling of alloy cluster expansions. In this thesis the alloy cluster expansion method is applied to study several different materials.
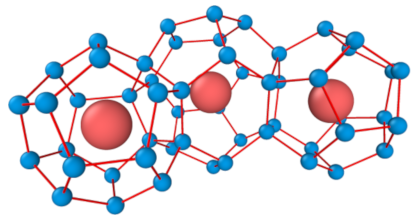
The first group of materials studied are inorganic clathrates. In paper II and III we studied the ordering behavior and related properties as a function of composition and temperature for the clathrates Ba8AlxSi46-x, Ba8AlxGe46-x, Ba8GaxGe46-x, and Ba8GaxSi46-x. We achieved very good agreement with the available experimental data for the site occupancy factors (SOFs).
In paper IV and V we constructed the phase diagram for the W-Ti and W-C system respectively. A cluster expansion for each system was constructed and the configurational free energy was calculated. By also including other contributions to the free energy, most notably the vibrational free energy, the phase diagrams for these systems could be constructed.
In paper VI we studied the SSZ-13 zeolite and showed both that the Löwenstein rule is not respected with hydrogen as counterion and provided a rationale for this behavior.