Machine Learning for Polaritonic Chemistry: Accessing chemical kinetics
C. Schäfer,
J. Fojt,
E. Lindgren,
and
P. Erhart
Journal of the American Chemical Society 146, 5402
(2024)
arXiv:2311.09739
doi: 10.1021/jacs.3c12829
zenodo: 10255268
(associated data)
Download PDF
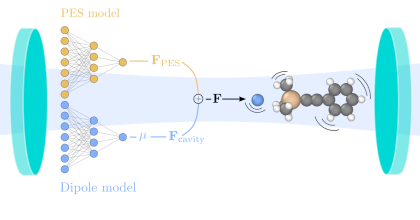
Altering chemical reactivity and material structure in confined optical environments is on the rise, and yet, a conclusive understanding of the microscopic mechanisms remains elusive. This originates mostly from the fact that accurately predicting vibrational and reactive dynamics for soluted ensembles of realistic molecules is no small endeavor, and adding (collective) strong light-matter interaction does not simplify matters. Here, we establish a framework based on a combination of machine learning (ML) models, trained using density-functional theory calculations, and molecular dynamics to accelerate such simulations. We then apply this approach to evaluate strong coupling, changes in reaction rate constant, and their influence on enthalpy and entropy for the deprotection reaction of 1-phenyl-2-trimethylsilylacetylene, which has been studied previously both experimentally and using ab initio simulations. While we find qualitative agreement with critical experimental observations, especially with regard to the changes in kinetics, we also find differences in comparison with previous theoretical predictions. The features for which the ML-accelerated and ab initio simulations agree show the experimentally estimated kinetic behavior. Conflicting features indicate that a contribution of electronic polarization to the reaction process is more relevant then currently believed. Our work demonstrates the practical use of ML for polaritonic chemistry, discusses limitations of common approximations and paves the way for a more holistic description of polaritonic chemistry.