Self-consistent calculations of charge self-trapping energies: A comparative study of polaron formation and migration in PbTiO3
E. Ghorbani,
L. Villa,
P. Erhart,
A. Klein,
and
K. Albe
Physical Review Materials 6, 30984
(2022)
doi: 10.1103/PhysRevMaterials.6.074410
Download PDF

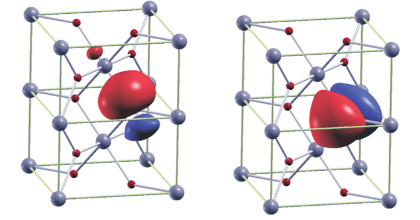
This study presents a systematic assessment of the behavior of self-trapped electrons in PbTiO3, which is a prototypical ferroelectric material with a wide range of technological applications. Since modeling of polarons depends sensitively on the applied method, the goal of this work is to identify the parameters used in density functional theory (DFT), which allow to predict the properties of polarons with high accuracy. The DFT+U method is employed to benchmark how the choice of k-mesh grids, lattice parameters, and pseudopotential (PP) affects the polaron trapping energy. Then, the appropriate parameters were used to study polaron trapping energy and its optical transition using the HSE06 hybrid functional. It is shown that the magnitude of the trapping energy is highly sensitive to the choice of the PP and the applied lattice parameters. A comparison of polaron trapping energies using the two functionals indicates proximity of the DFT+U result to the HSE06 result. Finally, configuration coordinate diagrams for the polaron-associated absorption and luminescence peaks in PbTiO3 are presented and compared to experiments.