Direct hot-carrier transfer in plasmonic catalysis
P. V. Kumar,
T. P. Rossi,
M. Kuisma,
P. Erhart,
and
D. J. Norris
Faraday Discussions 214, 189
(2019)
doi: 10.1039/c8fd00154e
Download PDF
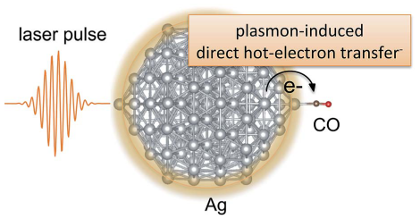
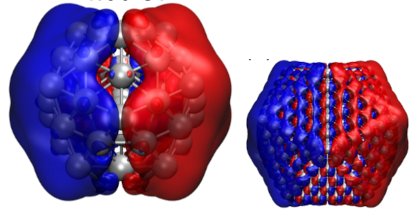
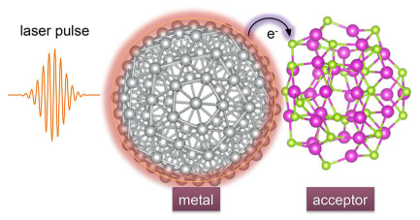
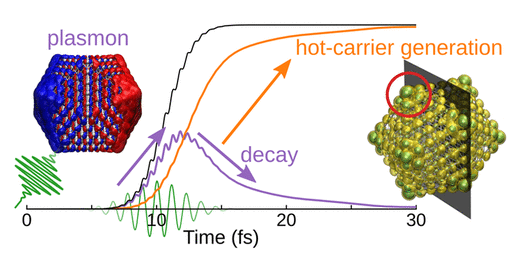
Plasmonic metal nanoparticles can concentrate optical energy and enhance chemical reactions on their surfaces. Plasmons can interact with adsorbate orbitals and decay by directly exciting a carrier from the metal to the adsorbate in a process termed the direct-transfer process. Although this process could be useful for enhancing the efficiency of a plasmonic reaction, it remains poorly understood. Here, we report a preliminary investigation employing time-dependent density-functional theory (TDDFT) calculations to capture this process at a model metal–adsorbate interface formed by a silver nanoparticle (Ag147) and a carbon monoxide molecule (CO). Direct hot-electron transfer is observed to occur from the occupied states of Ag to the unoccupied molecular orbitals of CO. We determine the probability of this process and show that it depends on the adsorption site of CO. Our results are expected to aid the design of more efficient metal–molecule interfaces for plasmonic catalysis.