Efficacy of the DFT+U formalism for modeling hole polarons in perovskite oxides
P. Erhart,
A. Klein,
D. Åberg,
and
B. Sadigh
Physical Review B 90, 14980
(2014)
arXiv:1402.0435
doi: 10.1103/PhysRevB.90.035204
Download PDF
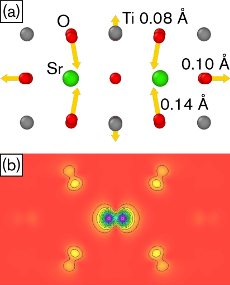
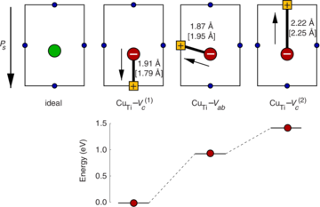
We investigate the formation of self-trapped holes (STH) in three prototypical perovskites (SrTiO3, BaTiO3, PbTiO3) using a combination of density functional theory (DFT) calculations with local potentials and hybrid functionals. First we construct a local correction potential for polaronic configurations in SrTiO3 that is applied via the DFT+U method and matches the forces from hybrid calculations. We then use the DFT+U potential to search the configuration space and locate the lowest energy STH configuration. It is demonstrated that both the DFT+U potential and the hybrid functional yield a piecewise linear dependence of the total energy on the occupation of the STH level, suggesting that self-interaction effects have been properly removed. The DFT+U model is found to be transferable to BaTiO3 and PbTiO3, and STH formation energies from DFT+U and hybrid calculations are in close agreement for all three materials. STH formation is found to be energetically favorable in SrTiO3 and BaTiO3 but not in PbTiO3, which can be rationalized by considering the alignment of the valence band edges on an absolute energy scale. In the case of PbTiO3 the strong coupling between Pb 6s and O 2p states lifts the valence band minimum (VBM) compared to SrTiO3 and BaTiO3. This reduces the separation between VBM and STH level and renders the STH configuration metastable with respect to delocalization (band hole state). We expect that the present approach can be adapted to study STH formation also in oxides with different crystal structures and chemical compositions.