First principles study of tungsten-based alloys: From defect thermodynamics to phase diagrams
L. Gharaee
Doctoral Thesis
(2017)
url: https://research.chalmers.se/publication/249363
Download PDF
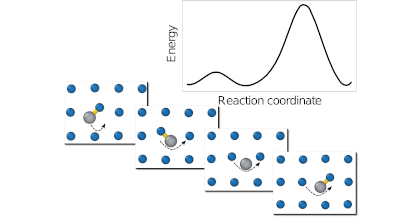
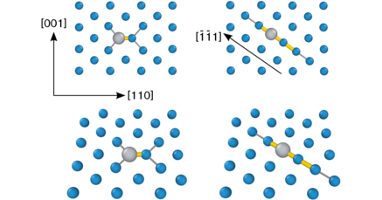
Nuclear fusion is a very attractive option for energy production as it is clean, safe and efficient. The major obstacle in construction of fusion power plants is the development of materials that can tolerate the extreme operational condition especially high-energy neutron flux. Tungsten is the prime candidate as armor material in fusion reactors due to its high strength and excellent high temperature properties. Tungsten alloys are interesting in this context because of the transmutation of tungsten to other elements upon neutron irradiation. In this thesis, properties of tungsten alloys were studied by means of first-principles calculations based on density functional theory (DFT). I investigated the thermodynamic and kinetic properties of intrinsic and extrinsic defects in tungsten. Ti, V and Re were shown to trap self-interstitials. Whereas mixed-interstitial migration was found to proceed via a non-dissociative mechanism with a lower barrier than that of vacancies. A closer look at trapping effects in W-Re, W-V, and W-Ti alloys showed that mixed-interstitials in all systems are strongly attracted to each other with large binding energies. They form interstitial pairs aligned along parallel first-neighbor ⟨111⟩ strings. This behavior is caused by the very large and anisotropic strain field of these interstitial defects. Low barriers for defect translation and rotation enable defect agglomeration and alignment even at moderate temperatures. Re-rich clusters that are observed in irradiated tungsten, at concentrations below the solubility limit, may eventually transform into intermetallic phases, which can affect materials hardness and embrittlement. To explain the formation of these precipitates, I carried out DFT calculations to characterize the landscape for diffusion. My results were subsequently used by collaborators to parametrize a kinetic Monte Carlo model. Simulations based on this model showed that both vacancy and interstitial mediated transports control cluster nucleation. Re-rich precipitates were seen to grow by attracting more mixed interstitials bringing solute atoms, which in turn attracted vacancies leading to recombination and solute agglomeration. The heat of mixing for the previously mentioned alloying elements was investigated further. For W-Ti, the equilibrium phase diagram below 1700 K is not known due to experimental difficulties. The present study revealed a negative heat of formation on the W-rich side, which is consistent with the experimentally observed asymmetric solubility. A revised solubility limit at low temperatures of the W-Ti phase diagram was derived on the basis of these calculations. Finally I assessed the performance of constraint-based functionals and considered the recently-developed consistent-exchange van der Waals density functional version vdW-DF-cx, the semi-local PBE and PBEsol functionals as well as the AM05 meta-functional. Structural and thermophysical properties of 3d, 4d, and 5d non-magnetic transition metals were computed at finite temperatures. It was shown that overall vdW-DF-cx provides an accurate description of thermophysical properties that is typically superior to the other functionals considered. It can thus be used to study systems that have both sparse and dense electron distributions.