Atomicrex - a general purpose tool for the construction of atomic interaction models
A. Stukowski,
E. Fransson,
M. Mock,
and
P. Erhart
Modelling and Simulation in Materials Science and Engineering 25, 23043
(2017)
doi: 10.1088/1361-651X/aa6ecf
Download PDF
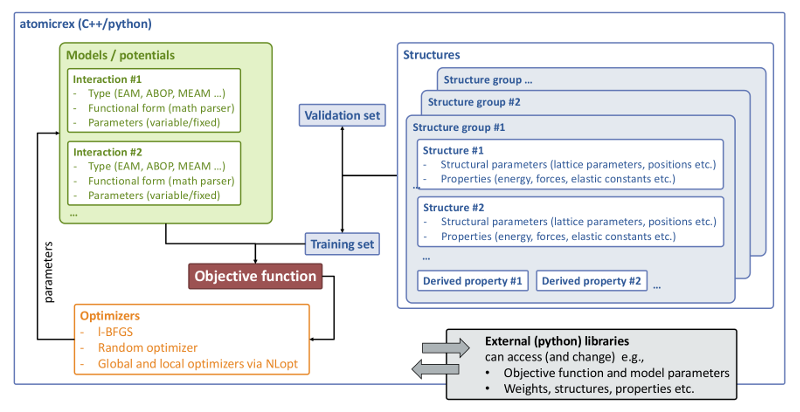
We introduce atomicrex, an open-source code for constructing interatomic potentials as well as more general types of atomic-scale models. Such effective models are required to simulate extended materials structures comprising many thousands of atoms or more, because electronic structure methods become computationally too expensive at this scale. atomicrex covers a wide range of interatomic potential types and fulfills many needs in atomistic model development. As inputs, it supports experimental property values as well as ab initio energies and forces, to which models can be fitted using various optimization algorithms. The open architecture of atomicrex allows it to be used in custom model development scenarios beyond classical interatomic potentials while thanks to its Python interface it can be readily integrated e.g., with electronic structure calculations or machine learning algorithms.