Understanding the Phase Diagram of Self-Assembled Monolayers of Alkanethiolates on Gold
J. Löfgren,
H. Grönbeck,
K. Moth-Poulsen,
and
P. Erhart
The Journal of Physical Chemistry C 120, 12059
(2016)
doi: 10.1021/acs.jpcc.6b03283
Download PDF
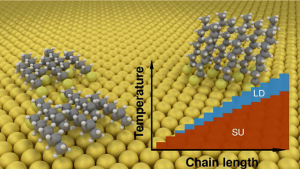
Alkanethiolate monolayers on gold are important both for applications in nanoscience as well as fundamental studies of adsorption and self-assembly at metal surfaces. While considerable experimental effort has been put into understanding the phase diagram of these systems, theoretical work based on density functional theory (DFT) has long been hampered by the inability of conventional exchange-correlation functionals to describe dispersive interactions. In this work, we combine dispersion-corrected DFT calculations using the new vdW-DF-CX functional with the ab initio thermodynamics method to study the stability of dense standing-up and low-coverage lying-down phases on Au(111). We demonstrate that the lying-down phase has a thermodynamic region of stability starting from thiolates with alkyl chains consisting of n≈3 methylene units. This phase emerges as a consequence of a competition between dispersive chain-chain and chain-substrate interactions, where the strength of the latter varies more strongly with n. A phase diagram is derived under ultra-high vacuum conditions, detailing the phase transition temperatures of the system as a function of the chain length. The present work illustrates that accurate ab initio modeling of dispersive interactions is both feasible and essential for describing self-assembled monolayers.