Electronic structure of LaBr3 from quasiparticle self-consistent GW calculations
D. Åberg,
B. Sadigh,
and
P. Erhart
Physical Review B 85, 125134
(2012)
arXiv:1201.386
doi: 10.1103/PhysRevB.85.125134
Download PDF
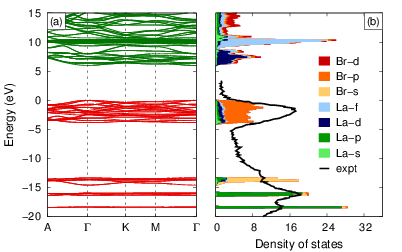
Rare-earth-based scintillators in general and lanthanum bromide (LaBr3) in particular represent a challenging class of materials due to pronounced spin-orbit coupling and subtle interactions between d and f states that cannot be reproduced by standard density functional theory (DFT). Here a detailed investigation of the electronic band structure of LaBr3 using the quasiparticle self-consistent GW (QPscGW) method is presented. This parameter-free approach is shown to yield an excellent description of the electronic structure of LaBr3. Specifically, it is able to reproduce the band gap, the correct level ordering and spacing of the 4f and 5d states, as well as the spin-orbit splitting of La-derived states. The QPscGW results are subsequently used to benchmark several computationally less demanding techniques including DFT+U, hybrid exchange-correlation functionals, and the G0W0 method. Spin-orbit coupling is included self-consistently at each QPscGW iteration and maximally localized Wannier functions are used to interpolate quasiparticle energies. The QPscGW results provide an excellent starting point for investigating the electronic structure of excited states, charge self-trapping, and activator ions in LaBr3 and related materials.