A first-principles study of helium storage in oxides and at oxide-iron interfaces
P. Erhart
Journal of Applied Physics 111, 113502
(2012)
doi: 10.1063/1.4707944
Download PDF
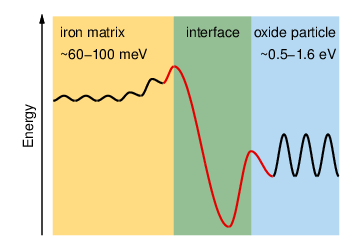
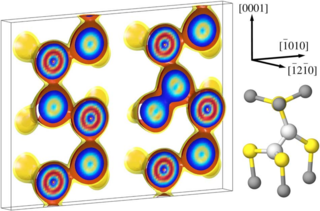
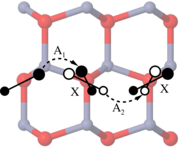
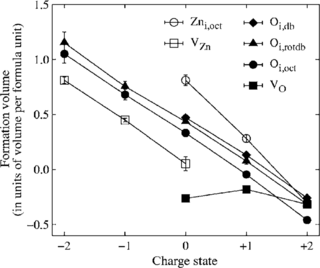
Density-functional theory calculations based on conventional as well as hybrid exchange-correlation functionals have been carried out to study the properties of helium in various oxides (Al2O3, TiO2, Y2O3, YAP, YAG, YAM, MgO, CaO, BaO, SrO) as well as at oxide-iron interfaces. Helium interstitials in bulk oxides are shown to be energetically more favorable than substitutional helium, yet helium binds to existing vacancies. The solubility of He in oxides is systematically higher than in iron and scales with the free volume at the interstitial site nearly independently of the chemical composition of the oxide. In most oxides, He migration is significantly slower and He-He binding is much weaker than in iron. To quantify the solubility of helium at oxide-iron interfaces two prototypical systems are considered (Fe-MgO, Fe-FeO-MgO). In both cases, the He solubility is markedly enhanced in the interface compared to either of the bulk phases. The results of the calculations allow to construct a schematic energy landscape for He interstitials in iron. The implications of these results are discussed in the context of helium sequestration in oxide dispersion strengthened steels, including the effects of interfaces and lattice strain.