First-principles study of intrinsic point defects in ZnO: role of band structure, volume relaxation and finite size effects
P. Erhart,
K. Albe,
and
A. Klein
Physical Review B 73, 205203
(2006)
doi: 10.1103/PhysRevB.73.205203
Download PDF
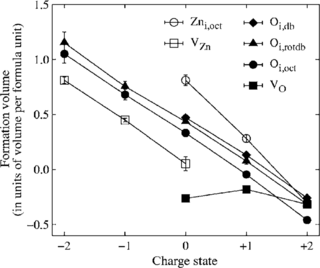
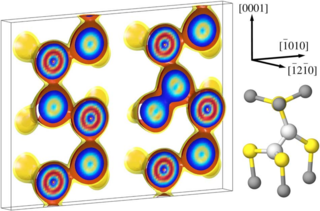
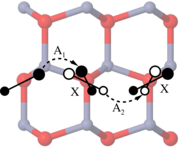
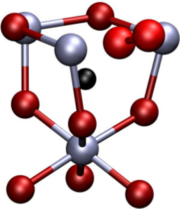
Density-functional theory (DFT) calculations of intrinsic point defect properties in zinc oxide were performed in order to remedy the influence of finite size effects and the improper description of the band structure. The generalized gradient approximation (GGA) with empirical self-interaction corrections (GGA+U) was applied to correct for the overestimation of covalency intrinsic to GGA-DFT calculations. Elastic as well as electrostatic image interactions were accounted for by application of extensive finite-size scaling and compensating charge corrections. Size-corrected formation enthalpies and volumes as well as their charge state dependence have been deduced. Our results partly confirm earlier calculations but reveal a larger number of transition levels: (1) For both, the zinc interstitial as well as the oxygen vacancy, transition levels are close to the conduction band minimum. (2) The zinc vacancy shows a transition rather close to the valence band maximum and another one near the middle of the calculated bandgap. (3) For the oxygen interstitials, transition levels occur both near the valence band maximum and the conduction band minimum.