First-principles study of the structure and stability of oxygen defects in zinc oxide
P. Erhart,
A. Klein,
and
K. Albe
Physical Review B 72, 085213
(2005)
doi: 10.1103/PhysRevB.72.085213
Download PDF
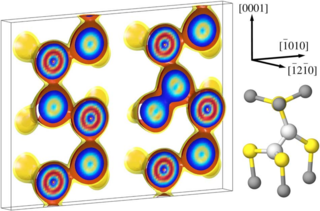
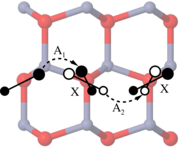
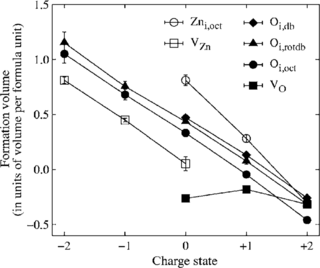
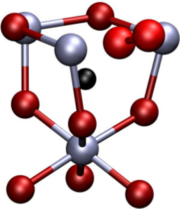
A comparative study on the structure and stability of oxygen defects in ZnO is presented. By means of first-principles calculations based on local density functional theory we investigate the oxygen vacancy and different interstitial configurations of oxygen in various charge states. Our results reveal that dumbbell-like structures are thermodynamically the most stable interstitial configurations for neutral and positive charge states due to the formation of a strongly covalent oxygen-oxygen bond. For negative charge states the system prefers a split-interstitial configuration with two oxygen atoms in almost symmetric positions with respect to the associated perfect lattice site. The calculated defect formation energies imply that interstitial oxygen atoms may provide both donor- and acceptor-like defects.