Ordering and segregation in the PdAuCu system: Bulk vs. surface
P. Tanner
Master′s Thesis
(2019)
doi: 20.500.12380/256745
Download PDF

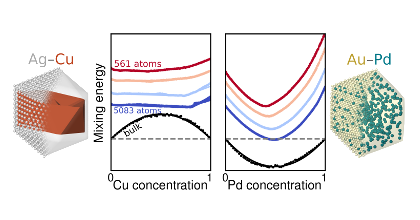
Nanoalloys are of interest in many fields of research such as catalysis, sensing and energy storage. They are, however, challenging to model due to the large number of available atomic configurations. In this thesis, the first-principle based method of cluster expansion is used to study atomic ordering and surface segregation of the PdAuCu system. It is shown that under vacuum conditions, Au shows a pronounced tendency to segregate towards the surface in relation to both Cu and Pd, while the CuPd system shows a slight excess of Pd at the surface. In addition, it is found that the AuCu system as well as the full AuCuPd system exhibit phase segregation in bulk due to several ordered phases, while AuPd and CuPd show complete miscibility.