Probing Glass Formation in Perylene Derivatives via Atomic Scale Simulations and Bayesian Regression
E. Lindgren,
J. Swensson,
C. Müller,
and
P. Erhart
The Journal of Physical Chemistry B 129, 6613
(2025)
arXiv:2501.15872
doi: 10.1021/acs.jpcb.5c00837
Download PDF
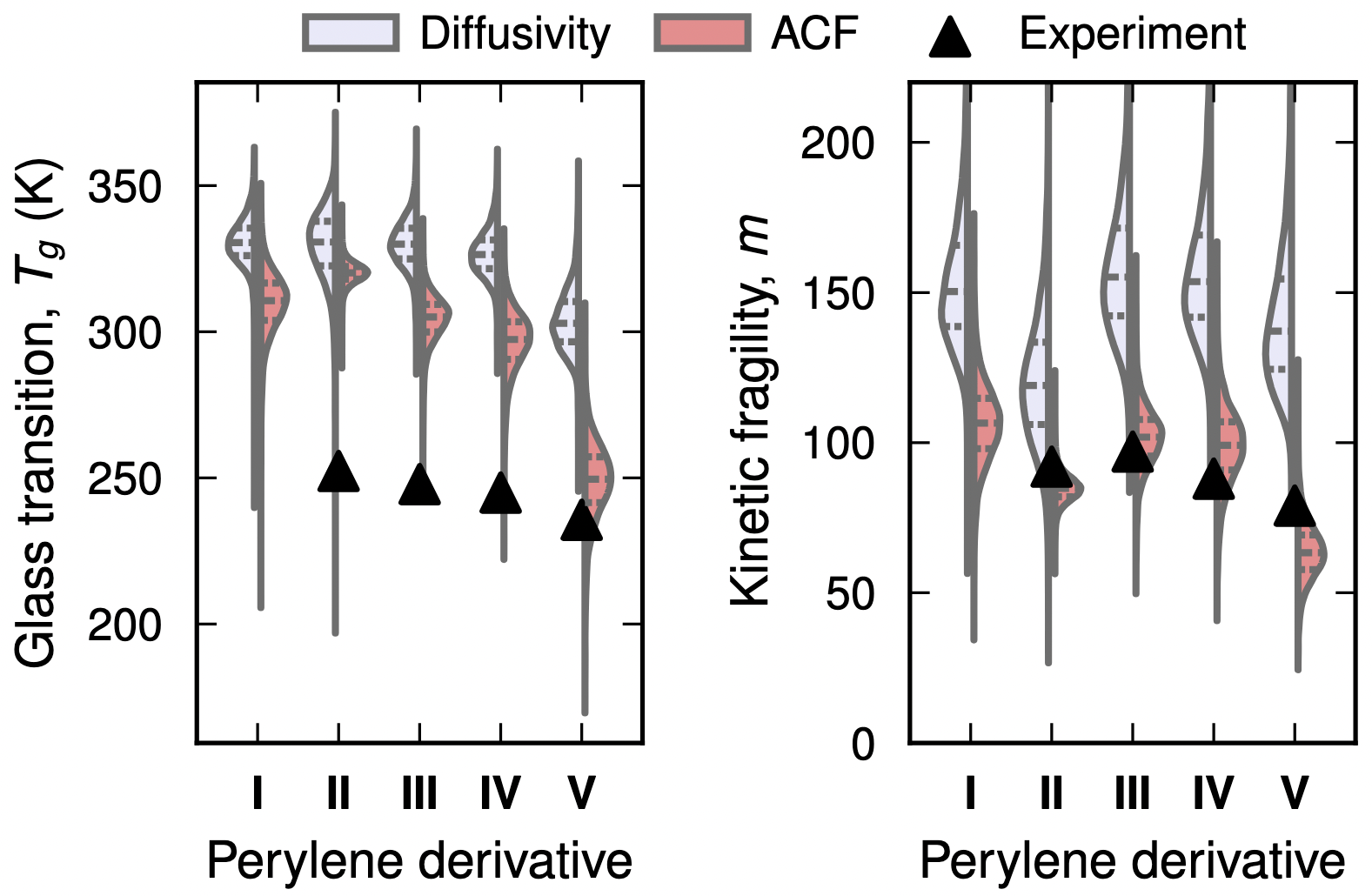
While the structural dynamics of chromophores are of interest for a range of applications, it is experimentally very challenging to resolve the underlying microscopic mechanisms. Glassy dynamics are also challenging for atomistic simulations due to the underlying dramatic slowdown over many orders of magnitude. Here, we address this issue by combining atomic scale simulations with autocorrelation function analysis and Bayesian regression, and apply this approach to a set of perylene derivatives as prototypical chromophores. The predicted glass transition temperatures and kinetic fragilities are in semi-quantitative agreement with experimental data. By analyzing the underlying dynamics via the normal vector autocorrelation function, we are able to connect the β and α -relaxation processes in these materials to caged (or librational) dynamics and cooperative rotations of the molecules, respectively. The workflow presented in this work serves as a stepping stone toward understanding glassy dynamics in many-component mixtures of perylene derivatives and is readily extendable to other systems of chromophores.