Quantifying Dynamic Tilting in Halide Perovskites: Chemical Trends and Local Correlations
J. Wiktor,
E. Fransson,
D. Kubicki,
and
P. Erhart
Chemistry of Materials 35, 6737
(2023)
arXiv:2304.07402
doi: 10.1021/acs.chemmater.3c00933
zenodo: 7889313
(associated data)
Download PDF
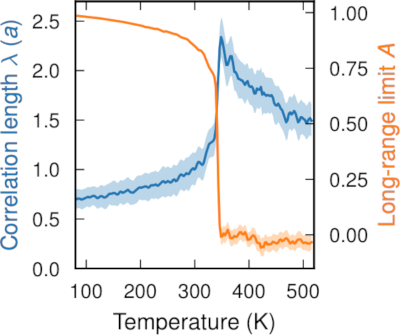
Halide perovskites have emerged as one of the most interesting materials for optoelectronic applications due to their favorable properties, such as defect-tolerance and long charge carrier lifetimes, which are attributed to their dynamic softness. However, this softness has led to apparent disagreements between the local instantaneous and global average structures of these materials. In this work, we rationalize this situation through an assessment of the local tilt angles of octahedra in the perovskite structure using large-scale molecular dynamics simulations based on machine-learned potentials trained using density functional theory calculations. We compare structural properties given by different density functionals (LDA, PBE, PBE+D3, PBEsol, SCAN, SCAN+rVV10, and vdW-DF-cx) and establish trends across a family of CsMX3 perovskites with M=Sn or Pb and X=Cl, Br, or I. Notably, we demonstrate strong short-range ordering in the cubic phase of halide perovskites. This ordering is reminiscent of the tetragonal phase and provides the bridge between the disordered local structure and the global cubic arrangement. Our results provide a deeper understanding of the structural properties of halide perovskites and their local distortions, which is crucial for further understanding their optoelectronic properties.