Phase transitions in inorganic halide perovskites from machine-learned potentials
E. Fransson,
J. Wiktor,
and
P. Erhart
The Journal of Physical Chemistry C 127, 13773
(2023)
arXiv:2301.03497
doi: 10.1021/acs.jpcc.3c01542
zenodo: 8014365
(associated data)
Download PDF
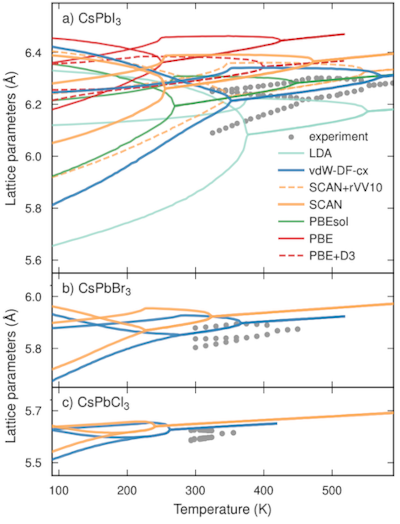
The atomic scale dynamics of halide perovskites have a direct impact not only on their thermal stability but their optoelectronic properties. Progress in machine-learned potentials has only recently enabled modeling the finite temperature behavior of these material using fully atomistic methods with near first-principles accuracy. Here, we systematically analyze the impact of heating and cooling rate, simulation size, model uncertainty, and the role of the underlying exchange-correlation functional on the phase behavior of CsPbX3 with X=Cl, Br, and I, including both the perovskite and the delta-phases. We show that rates below approximately 60 K/ns and system sizes of at least a few ten thousand atoms should be used to achieve convergence with regard to these parameters. By controlling these factors and constructing models that are specific for different exchange-correlation functionals we then assess the behavior of seven widely used semi-local functionals (LDA, vdW-DF-cx, SCAN, SCAN+rVV10, PBEsol, PBE, and PBE+D3). The models based on LDA, vdW-DF-cx, and SCAN+rVV10 agree well with experimental data for the tetragonal-to-cubic-perovskite transition temperature in CsPbI3, and also achieve reasonable agreement for the perovskite-to-delta phase transition temperature. They systematically underestimate, however, the orthorhombic-to-tetragonal transition temperature. All other models, including those for CsPbBr3 and CsPbCl3, predict transition temperatures below the experimentally observed values for all transitions considered here. Among the considered functionals vdW-DF-cx and SCAN+rVV10 yield the closest agreement with experiment, followed by LDA, SCAN, PBEsol, PBE, and PBE+D3. Our work provides guidelines for the systematic analysis of dynamics and phase transitions in inorganic halide perovskites and similar systems. It also serves as a benchmark for the further development of machine-learned potentials as well as exchange-correlation functionals.