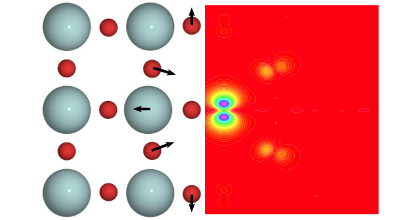
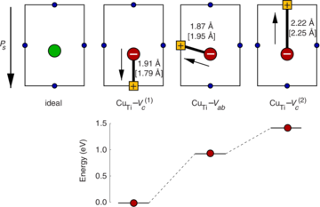
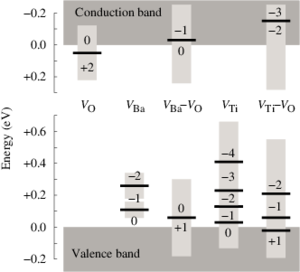
Implications of the band gap problem on oxidation and hydration in acceptor-doped barium zirconate
A. Lindman,
P. Erhart,
and
G. Wahnström
Physical Review B 91, 245114
(2015)
doi: 10.1103/PhysRevB.91.245114
Download PDF
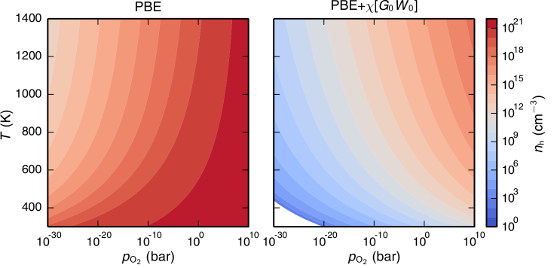
Charge carrier concentrations in acceptor-doped proton-conducting perovskites are to a large extent determined by the hydration and oxidation of oxygen vacancies, which introduce protons and holes, respectively. First-principles modeling of these reactions involves calculation of formation energies of charged defects, which requires an accurate description of the band gap and the position of the band edges. Since density-functional theory (DFT) with local and semilocal exchange-correlation functionals (LDA and GGA) systematically fails to predict these quantities this can have serious implications on the modeling of defect reactions. In this study we investigate how the description of band gap and band-edge positions affects the hydration and oxidation in acceptor-doped BaZrO3. First-principles calculations are performed in combination with thermodynamic modeling in order to obtain equilibrium charge carrier concentrations at different temperatures and partial pressures. Three different methods have been considered: DFT with both semilocal (PBE) and hybrid (PBE0) exchange-correlation functionals, and many-body perturbation theory within the G0W0 approximation. All three methods yield similar results for the hydration reaction, which are consistent with experimental findings. For the oxidation reaction, on the other hand, there is a qualitative difference. PBE predicts the reaction to be exothermic, while the two others predict an endothermic behavior. Results from thermodynamic modeling are compared with available experimental data, such as enthalpies, concentrations, and conductivities, and only the results obtained with PBE0 and G0W0, with an endothermic oxidation behavior, give a satisfactory agreement with experiments.