Analytical Potential for Atomistic Simulations of Silicon and Silicon Carbide
P. Erhart
and
K. Albe
Physical Review B 71, 14985
(2005)
doi: 10.1103/PhysRevB.71.035211
Download PDF
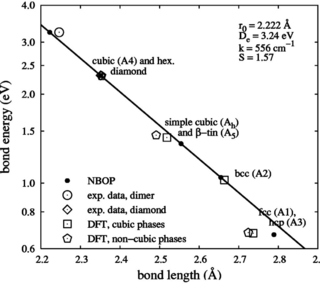
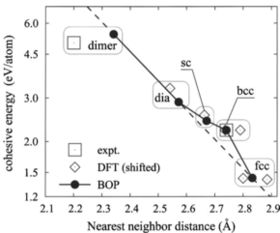
We present an analytical bond-order potential for silicon, carbon and silicon carbide that has been optimized by a systematic fitting scheme. The functional form is adopted from a preceeding work [Phys. Rev. B. 65, 195124 (2002)] and is built on three independently fitted potentials for Si-Si, C-C and Si-C interaction. For elemental silicon and carbon, the potential perfectly reproduces elastic properties and agrees very well with first-principles results for high-pressure phases. The formation enthalpies of point defects are reasonably reproduced. In case of silicon stuctural features of the melt agree nicely with data taken from literature. For silicon carbide the dimer as well as the solid phases B1, B2 and B3 were considered. Again, elastic properties are very well reproduced including internal relaxations under shear. Comparison with first-principles data on point defect formation enthalpies shows fair agreement. The successful validation of the new potentials for configurations ranging from the molecular to the bulk regime, indicates the transferability of the potential model and makes it a good choice for atomistic simulations that sample a large configuration space.