The role of thermostats in modeling vapor phase condensation of silicon nanoparticles
P. Erhart
and
K. Albe
Applied Surface Science 226, 12
(2004)
doi: 10.1016/j.apsusc.2003.11.003

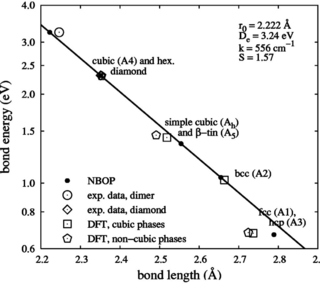
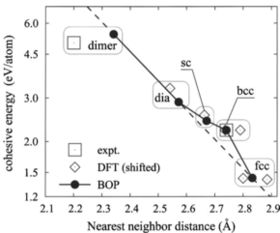
Condensation of silicon nanoclusters from the vapor phase is investigated by means of atomic scale molecular dynamics simulations and discussed with respect to its sensitivity to thermodynamic boundary conditions. For systems consisting of the condensing species only thermostats homogeneously acting on all atoms are unable to mimic the role of a background gas. Only if the inert gas is added to the simulation volume a realistic description of vapor phase condensation is possible. In this paper, we discuss appropriate methods to thermalize the background gas by comparing the effect of different thermostats on nanocluster formation. Moreover, we study the influence of the interatomic potential describing the interaction of inert atoms and reactive species.