Dipolar coupling of nanoparticle-molecule assemblies: An efficient approach for studying strong coupling
J. Fojt,
T. P. Rossi,
T. J. Antosiewicz,
M. Kuisma,
and
P. Erhart
Journal of Chemical Physics 154, 094109
(2021)
arXiv:2101.05160
doi: 10.1063/5.0037853
Download PDF
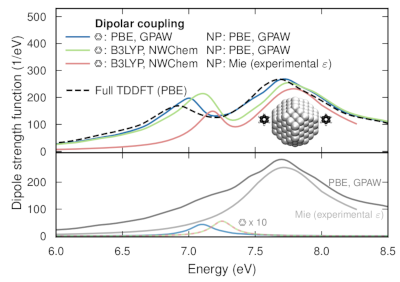
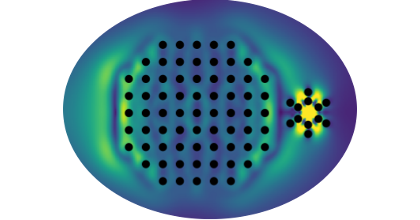
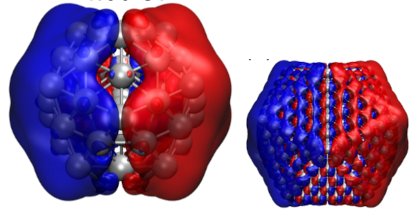
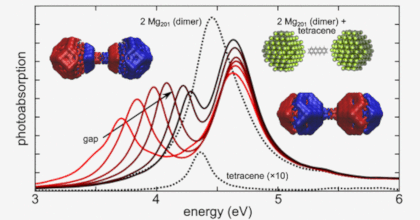
Strong light-matter interactions facilitate not only emerging applications in quantum and non-linear optics but also modifications of materials properties. In particular the latter possibility has spurred the development of advanced theoretical techniques that can accurately capture both quantum optical and quantum chemical degrees of freedom. These methods are, however, computationally very demanding, which limits their application range. Here, we demonstrate that the optical spectra of nanoparticle-molecule assemblies, including strong coupling effects, can be predicted with good accuracy using a subsystem approach, in which the response functions of the different units are coupled only at the dipolar level. We demonstrate this approach by comparison with previous time-dependent density functional theory calculations for fully coupled systems of Al nanoparticles and benzene molecules. While the present study only considers few-particle systems, the approach can be readily extended to much larger systems and to include explicit optical-cavity modes.